
项目方案
一、 研究目的
采用随机对照试验设计,验证pBFS指导下的TMS干预方法对中重度自闭症患者社会功能改善的有效性和安全性。
二、 研究设计
大样本、前瞻性随机对照试验设计。
三、 研究场所与对象
研究场所:北京博爱医院及北京优脑银河诊所。
研究对象:通过社区招募的中重度自闭症患者。
四、 研究期限
自委托书任务协议书签署起至签署后一年止。
五、 技术路线图
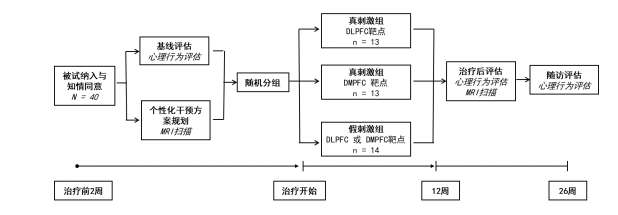
六、 研究实施
(一) 研究对象
1、诊断标准
通过专业的精神科医生或临床心理医生评估,符合DSM-IV或DSM-V关于自闭症的诊断标准,程度为中重度,明显影响社会功能。
2、纳入标准
a) 年龄6-30岁;
b) ADOS和ADI-R的评估结果达到自闭症标准切截线
c) 患儿/者的监护人自愿参与治疗,能够配合完成治疗并签署知情同意书
3、排除标准
a) 有严重视觉、听觉或运动障碍不能配合检查和治疗;
b) 患有严重肝肾功能障碍或其他严重躯体疾病;
c) 有癫痫、脑外伤等神经系统疾病或病史;
d) 并发精神分裂症、双向情感障碍等精神错乱症状;
e) 体内有金属异物或其他植入体内的任何电子装备者。
4、剔除标准
a) 在基线测评时,病情变化不再符合纳入标准者;
b) 试验过程中,因其他疾病需要中断治疗者;
c) 试验过程中,患者心理行为剧烈表现,如出现自伤或自杀行为。
d) 试验过程中病情明显加重,经主治医师评估后退出此项研究。
5、退出、脱落病例
因以下原因未完成临床方案的入组病例应视为脱落:
5.1 失访;
整个治疗过程中缺席治疗>6次
5.2病人自行退出。
患者及家属要求终止试验,经实验者解答疑虑后依然决定退出。
6、样本量
计划入组40名患者,以满足试验中的统计要求。
(二) 研究方法
1、分组方法
本试验采用分层随机方法(分层因素:年龄、ADOS基线分数),以1:1:1的比例随机分配DLPFC靶点组、DMPFC靶点组和假刺激组进行12周的治疗。真刺激组每组13人,假刺激组14人,共40人。
2、盲法
本试验采取双盲设计。患者、患者家属及陪护人员、临床医护人员,心理行为评估师,TMS操作员,行为干预师,研究者在数据收集及分析完成前均不知道分组类型。有专门的研究者生成随机序列,分组和设盲,该研究者不参与其他任何过程。
心理行为评估师、TMS操作员与行为干预师不重复。
(三) 干预措施
1、基础治疗
如纳入患者有长期服用的药物或干预项目,应继续保持,并进行记录。
2、各组干预措施
2.1 设备情况
由CPNL统一确定导航和刺激设备;线圈:配套8字线圈+假刺激线圈;
2.2静息运动阈值 (RMT) 测定
将8字线圈的中心位置放于右手拇指对应的大脑皮质运动区的头皮处,嘱患者完全放松,逐渐调节经颅磁刺激输出强度,连续10次单刺激中能引出5次运动诱发电位 (MEP) 波幅大于50μV的最小刺激强度,或10次刺激中至少有5次引起右手拇指肌肉收缩的最小强度
2.3 DLPFC靶点组和DMPFC靶点组
根据个体脑功能图谱确定位于DLPFC或DMPFC的个性化靶点。使用CPNL提供的导航系统精确定位靶点和实施刺激。刺激强度为100% RMT,丛内频率50Hz,脉冲3个,丛间频率5Hz,刺激2s,间隔8s,共600s,总脉冲为1800个。
2.4 假刺激组
根据个体脑功能图谱确定位于DLPFC/DMPFC的个性化靶点。使用导航系统精确定位靶点。使用假线圈,不输出磁场。强度为100% RMT,丛内频率50Hz,脉冲3个,丛间频率5Hz,刺激2s,间隔8s,共600s,总脉冲为1800个。
3、疗程
周一至周五工作日每日治疗3次,每次约9分钟(1800个脉冲),治疗间隔为1小时10分钟,共治疗12周。在整个治疗过程中,同一病人每日均在上午或下午治疗,且每次治疗使用同一台刺激设备。
每天治疗间隔由行为干预师给予四次30分钟的行为干预课程。
(四) 观察指标及观察时点
1、基线指标
1.1人口学指标:性别、年龄、民族、身高、体重、受教育程度、婚姻情况;
1.2生命体征:体温、脉搏、呼吸、血压;
1.3疾病诊断相关情况:发病年龄、病程、分娩方式、既往和现在的治疗情况 (包括用药情况和行为评估)等;
1.4 MRI数据采集
为了完成扫描,所有患者需要在麻醉情况下进行MRI数据采集,采集患者的结构和静息态功能磁共振数据。
项目中使用同一台MRI设备,具体参数由CPNL根据研究承担单位设备确定。基本条件包括,使用32通道头部线圈。结构相数据满足空间分辨率1毫米,覆盖整个头部。静息态数据满足3毫米左右空间分辨率,2至3秒时间分辨率,覆盖全脑 (包括全部小脑)。采集4个8分钟序列,共32分钟静息态功能磁共振数据。
1.5心理行为评估量表:
有资质的专业人员(医生或心理学家)对患者进行行为评估。确保每个被试在每个时间点的评估人员相同,且与TMS治疗师及行为干预师不同。评估内容包括:
自闭症诊断观察量表 (Autism Diagnostic Observation Schedule,ADOS)
孤独症诊断面谈-修订版(Autism Diagnostic Interview–Revised,ADI-R)
2、临床观察指标及时点
2.1 评价指标:
在治疗结束后(第13周)和治疗后随访(26周)中,由同一评估师对患者进行行为评估,评估内容包括ADOS,ADI-R。
治疗结束后采集患者麻醉状态下的结构和静息态功能磁共振数据。采集参数与基线相同。
2.1.1 主要结局指标
治疗前后ADOS总评分的改变;
2.1.2 次要结局指标
1)治疗前后ADOS分项目评分的改变;
3)随访时ADOS总评分的改变
2.1.3 其他指标
1)在随访时ADOS分项目评分相较于治疗前的改变;
2)治疗前后脑功能连接与脑结构的改变。
2.2 基础用药及其他治疗:记录研究期间患者所有用药情况,并记录用药使用原因、药名、用量和使用持续时间;同时记录患者所有的特殊教育和干预项目及其频率、强度与持续时间。
2.3不良事件及不良反应:记录研究期间患者出现的所有不良事件及不良反应,并记录处理过程。
2.4病例脱出或剔除:记录研究期间出现的病例脱出或剔除情况。
(五) 访视方式
1. 随访期内:随访至干预结束后的14周,门诊复诊,评估患者的心理行为表现。
时期 | 筛查期 | 干预前 | 干预期 | 干预后 | 随访 |
日期(天/月) |
| -3~0天 | 0-12周 | 13周 | 26周 |
知情同意 | X |
|
|
|
|
人口资料 | X |
|
|
|
|
MRI扫描 | X |
|
| X |
|
随机分组 |
| X |
|
|
|
行为评估 |
| X |
| X | X |
TMS干预 |
|
| X |
|
|
盲法评估 |
|
|
| X |
|
不良事件 |
|
|
| X | X |
表一 流程时刻表
(六) 退出、脱落和终止试验的规定
所有签署了知情同意书并筛选合格进入试验的受试者,均有权随时退出临床试验,无论何时何因退出,只要没有完成临床试验全程观察,均为脱落病例。
退出、脱落病例的处理:当受试者脱落后,研究者必须在病例报告表(Case Report From,CRF)中填写脱落原因,并尽可能与受试者联系,完成所能完成的评估项目,并填写治疗末访视记录表。对因不良反应而脱落,经访视最后判断与试验有关者,必须记录在CRF中并通知承担单位。退出、脱落病例应进行报告和分析。
发生下列情况时受试者应中止试验:
1. 治疗期间出现突发性生理疾病,心理行为剧烈改变,如出现自伤或自杀行为;
2. 治疗期间病情加重,经主治医师评估后建议退出;
3. 受试者因个人原因不再配合研究者安排,或临床医师评估受试者依从性差,不适合进行研究;
4. 治疗期间患者因其他原因无法继续完成治疗
中止研究的病例应记录中止的时间、原因,并进行报告和分析。
(七) 安全性观察
严重不良事件及时向研究中心负责人、课题组负责人及联系人报告。所有不良事件均做出与研究关联性的评价。
(八) 不良事件记录、处理与报告
1.不良事件是指受试者在本次临床研究中发生的任何不良医学事件,无论这一事件与上述使用的研究措施是否有因果关系。临床研究过程中研究者仍需记录所有观察到、经非诱导性提问得到的或受试者主诉的不良事件,不论不良事件应发生在哪个组,也不论不良事件是否与治疗方案相关。同时,也应报告研究期间新发生的疾病或原有症状加重情况。临床干预效果不佳不应被记录为不良事件。
2.TMS(涉及的干预措施)常见不良反应包含且不限于以下情况:
2.1 一般不良反应:头痛;刺激部位头皮不适;面部肌肉刺痛、痉挛或抽搐;头晕目眩;疲劳等。
2.2罕见不良反应:诱发癫痫;耳鸣、听力下降等。
2.3其他不可预见的意外情况。
3.不良事件的记录与报道 对试验期间出现的不良反应,临床医师要详细记录于观察表,并评价不良反应与TMS及药物的相关性,及与合并药物的相互作用,记录所出现的意外情况,将其症状或疾病的程度、发病日期、频率、持续时间、缓解日期、处理措施、治疗经过、结果及随访情况等记录于病例观察表上,并且在综合考虑合并疾病、合并用药/方法的基础上,评价其与试验的相关性,并由医师详细记录,并注明该病例是否继续进行试验等情况,由研究者签名。
4.不良事件的上报 在试验中出现严重不良事件,应在24小时内口头通知申办者、伦理委员会和组长单位,并填写“严重不良事件报告表”,申办者将立即通知各参研医院并保证满足所有法律法规要求的报告程序。
5.不良事件的处理 发现不良反应时,观察医师可根据病情决定是否中止观察。出现严重不良事件,承担临床试验的单位须立即采取必要处理措施,保护受试者安全。所有不良事件都应当追踪调查,详细记录处理经过及结果,直到得到妥善解决或病情稳定,若化验异常者应追踪至恢复正常。追踪随访方式可以根据不良反应的轻重选择住院、门诊、家访、电话、通讯等多种形式。
(九) 患者的依从性及盲法测评
1. 在治疗过程中,需要通过量表或医师问询的方式统计患者服药和治疗情况。每周记录一次。
2. 治疗次数可用于评测患者依从性,指标每周记录一次。根据试验方案要求,需保证患者的依从性≥90%。
依从性= (受试者治疗次数/预定治疗次数)×100%
3. 在治疗结束后,临床实验协调员应对患者或患者家属、临床医师、治疗师、评估师进行量表或问询,让各角色猜测患者分组,并报告猜测原因,以考察盲法的有效性。
(十) 统计分析
1. 数据清理
缺失值:剔除缺失值超过20%的被试。对缺失值使用差值法进行补全。
极端值:剔除3个标准差外的数据。
2. 随机分组测评
为确认治疗前试验组和控制组在人口学、基线评估指标上是否存在差异,根据数据特征分别使用卡方检验,秩和检验或独立样本t检验。若发现显著差异,该变量将作为协变量在相应的有效性分析中加以控制。
3. 有效性分析
根据数据分布状态,采用独立样本t检验或秩和检验考察真刺激组和假刺激组,以及不同靶点间在干预后行为指标的变化。
采用配对t检验考察真刺激组和假刺激组以及不同靶点干预组内治疗前后的脑功能变化。
3. 安全性分析
报告各组不同种类不良事件次数/频率。若单类不良事件达到5次以上,使用卡方检验考察不良事件的次数/频率与干预类型的关系,并使用相关分析考察不良事件的次数/频率与患者人口学、基线评估指标的关系。
七、 数据管理
临床试验机构和研究者应当确保临床试验所形成数据、文件和记录的真实、准确、清晰、安全。
(一) CRF记录及核查
CRF表由研究者填写,每个入选病例必须完成CRF表及相关数据录入。完成的CRF表由监查员审查后,移交数据管理员,进行管理工作。最后结果由研究项目申办方进行审核。
观察医生在填写CRF表时,应注意以下几点:
1、全部病例均按以研究方案进行观察、治疗,按照 CRF 填写要求认真填写。
2、如实记录受试者用药情况、合并症情况。
3、病历及CRF表作为原始记录,不得更改,做任何更正时不得改变原始记录,只能采用附加叙述说明理由,由参加临床研究的医师签名并注明日期。
临床协作单位设置专人负责CRF的质量控制;CRF的录入、核对需两名人员分别进行。在盲态审核并确认建立的数据库正确后,由主要研究者、数据管理人员和统计分析人员对数据进行锁定。
(二) 影像数据管理
影像数据采集和收集:每例影像数据的采集都应该获得数据的原始DICOM文件并在CRF中按扫描顺序如实记录扫描序列和扫描过程中可能影响数据质量的相关事件,并根据临床协作单位设备情况,将每例数据导出到光盘或移动硬盘中。脱敏数据移交到研究项目申办方进行数据质量控制和备份;
影像数据的质量控制:每例影像数据及时移交到研究项目申办方进行质量控制,并告知临床协作单位数据质量是否合格和否需要重新扫描;
影像数据的备份:每例影像数据在临床协作单位进行一次备份,在研究项目申办方进行三重备份。
(三) 数据疑问与答疑
对病例报告表中存在的疑问,监查人员、数据管理员将填写疑
问解答表 (DRQ),并通过监查员向研究者发出询问,研究者应尽快解答并返回,监查人员、数据管理员根据研究者的回答进行数据修改、确认与录入,必要时可以再次发出DRQ。
八、 质量控制
(一) 制定随机对照试验实施的SOP;
(二)对参与临床研究的人员统一进行方法学和相关操作程序的培训,培训内容包括临床研究方案、临床诊断标准、TMS治疗方案、量表评估、CRF表填写等;
(三)随机抽取20%~50%患者的CRF与原始病历进行核对;
(四)质量控制人员定期对进行现场监查,建议至少在项目起始、中期及末期分三次进行。
九、 总结报告与资料保管
研究者应保存受试者所有的详细原始记录,记录的数据应保证完整、及时、清晰。CRF表、原始记录等应清楚、详细并易被参加此临床研究的人员辨识。CRF表及研究相关所有记录材料由专人保存,研究医院保存正在进行的CRF表,已完成CRF表需送交课题承担单位进行存档。
研究者至少须在CRF表的完成页签名,以证实所有数据的准确性和合法性。CRF表及原始记录只能由研究者进行修改。对CRF表及原始记录的任何修改都不得将原始数据涂抹掉。正确的修改方法是在原数据上划线,再将修改后的数据写在原始数据旁边,并签署日期及修改人员的姓名缩写。若改动不清楚则应说明原因。若研究者调动工作或不再履行其研究职责,须将替换医生的资质情况通知组长单位。
临床研究者及申办方应定期进行临床试验的安全与质量控制总结报告,针对试验中存在的问题及不稳定因素进行说明,在不影响整体实验方案的基础上提出改进意见,以提高实验进程效率并保障研究质量。
要求以下资料完整,归档及时,由后勤组专人专柜保管:试验方案及更新、研究者手册及更新、伦理资料及伦理委员会批文、知情同意书、CRF表、试验原始资料(病历、实验室检查等)、试验相关SOP、试验质量控制相关文件:监查计划、质控报告、监查报告等。